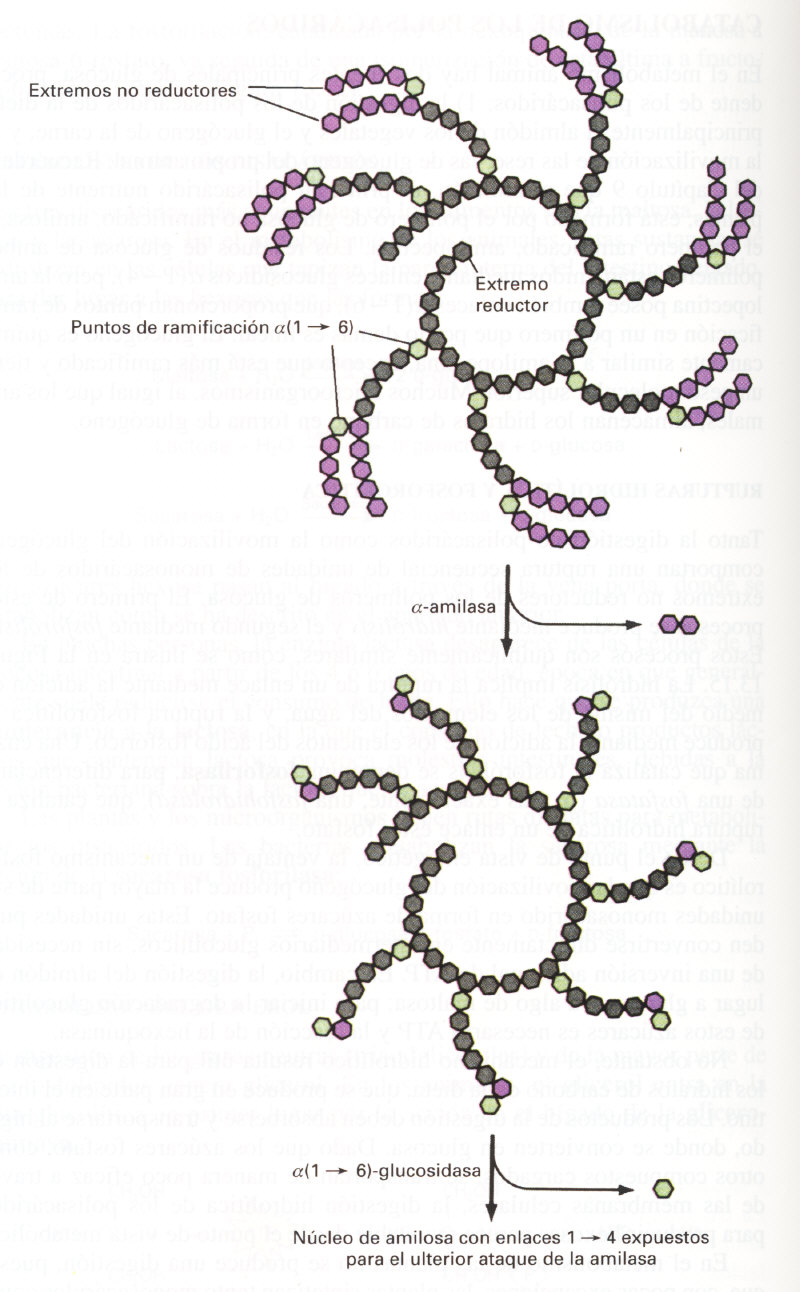
1.1. Digestión de Almidón y Glucógeno:
En los animales, la digestión del almidón y del glucógeno empieza en la , con la acción de la alfa-amilasa que se secreta en la saliva. Esta enzima rompe con los enlaces internos alfa 1-4 de ambos polímeros. En el intestino, la digestión continúa, facilitada por la alfa- amilasa secretada por el páncreas. Esta enzima degrada la amilosa a maltosa y un poco de glucosa. Sin embargo, solo degrada parcialmente la amilopectina y el glucógeno, porque no es capaz de romper los enlaces alfa 1,6 que se encuentran en los puntos de ramificación. El producto de la digestión completa de la amilopectina o del glucógeno por la alfa-amilasa se denomina dextrina límite, para continuar su degradación es necesaria la acción de una "enzima desramificante", la alfa 1-6 glucosidasa (también llamada isomaltasa). Esta acción expone un nuevo grupo de ramificaciones con enlaces alfa 1-4, que pueden ser atacadas por la alfa-amilasa, hasta alcanzar una nueva serie de ramificaciones con enlaces alfa 1-6.
El resultado final de la acción secuencial de estas dos enzimas es la degradación completa del almidón o glucógeno a maltosa y algo de glucosa. La maltosa se rompe hidrolíticamente por la maltasa, dando 2 moléculas de glucosa, que se absorbe a continuación al torrente circulatorio y se transporta a los diversos tejidos para su utilización. (7)
___________
- Resumen:
- Importancia Biomédica del Glucógeno:
- Catabolismo del Glucógeno (glucogenólisis):
- Cascada amplificadora de la degradación del glucógeno, estimulada por adrenalina:
- Funciones de las reservas de glucógeno en el músculo y el hígado:
- Fisiopatologías del metabolismo del glucógeno:
- Bibliografia
Los signos y síntomas clínicos más característicos son: hepatomegalia, hipoglucemia, osteoporosis, entre otros y en ocasiones son débiles y poco aparentes. Existen diversas pruebas de laboratorio para realizar un diagnóstico específico.
Palabras clave:
Glucógeno, glucostato, enzimas, cascada amplificadora de degradación de glucógeno, glucogenosis.
Introducción:
El glucógeno es la forma principal de almacenaje de carbohidratos en los , se encuentra en proporción en el hígado (hasta 6%) y en el músculo, donde rara vez excede de 1%. Sin embargo, debido a su masa mayor, el músculo almacena tres a cuatro veces la cantidad de glucógeno que tiene el hígado como reserva. Al igual que el almidón, es un polímero ramificado de alfa-glucosa .(9)
En las células hepáticas, el glucógeno aparece en forma de grandes gránulos, constituidos por agrupaciones de simples moléculas, muy ramificadas, por lo que tiene un peso molecular muy elevado. A semejanza de la amilopectina, el glucógeno es un polisacárido de la D-glucosa con enlaces alfa 1-4, sin embargo, está más ramificado, y su molécula es más reducida que la amilopectina; las ramificaciones aparecen cada 8 a 12 residuos de glucosa.
El glucógeno puede aislarse de los tejidos animales digiriéndolos con disoluciones calientes de KOH en las que los enlaces no reductores alfa 1-4 y alfa 1-6 son estables. (5)
Importancia Biomédica del Glucógeno:
La función del glucógeno muscular es actuar como una fuente de fácil disponibilidad de unidades de hexosa para la glucólisis dentro del propio músculo. El glucógeno hepático sirve en gran parte para exportar unidades de hexosa para la conservación de la glucosa sanguínea, en particular entre comidas.
Después de 12 a 18 horas de ayuno, el hígado agota su reserva de glucógeno. El glucógeno muscular sólo disminuye de manera significativa después de ejercicio vigoroso prolongado. Puede inducirse un almacenaje mayor de glucógeno muscular con dietas ricas en carbohidratos después de la depleción por el ejercicio. Las "enfermedades por almacenamiento de glucógeno" son un grupo de trastornos hereditarios que se caracterizan por movilización deficiente del glucógeno y depósito de formas anormales del mismo, conduciendo a debilidad muscular e inclusive muerte. (9).
Molécula de Glucógeno:

(9) Murray, Robert K., Granner, Daryl K., Mayes, Peter, A. ,Rodwell, Víctor W.F. "Bioquímica de Harper". 1992. 14a. edición. Editorial: El Manual Moderno. 132
1. Metabolismo Bioquímico del Glucógeno
1.1. Digestión de Almidón y Glucógeno:
En los animales, la digestión del almidón y del glucógeno empieza en la , con la acción de la alfa-amilasa que se secreta en la saliva. Esta enzima rompe con los enlaces internos alfa 1-4 de ambos polímeros. En el intestino, la digestión continúa, facilitada por la alfa- amilasa secretada por el páncreas. Esta enzima degrada la amilosa a maltosa y un poco de glucosa. Sin embargo, solo degrada parcialmente la amilopectina y el glucógeno, porque no es capaz de romper los enlaces alfa 1,6 que se encuentran en los puntos de ramificación. El producto de la digestión completa de la amilopectina o del glucógeno por la alfa-amilasa se denomina dextrina límite, para continuar su degradación es necesaria la acción de una "enzima desramificante", la alfa 1-6 glucosidasa (también llamada isomaltasa). Esta acción expone un nuevo grupo de ramificaciones con enlaces alfa 1-4, que pueden ser atacadas por la alfa-amilasa, hasta alcanzar una nueva serie de ramificaciones con enlaces alfa 1-6.
El resultado final de la acción secuencial de estas dos enzimas es la degradación completa del almidón o glucógeno a maltosa y algo de glucosa. La maltosa se rompe hidrolíticamente por la maltasa, dando 2 moléculas de glucosa, que se absorbe a continuación al torrente circulatorio y se transporta a los diversos tejidos para su utilización. (7)
1.2. Movilización del Glucógeno:
Las principales reservas de glucógeno de los vertebrados se encuentran en el músculo esquelético y en el hígado. La degradación de estas reservas en energía utilizable, o movilización del glucógeno, requiere las rupturas fosforolíticas secuenciales de los enlaces alfa 1-4, catalizadas por la glucógeno fosforilasa. En las plantas, el almidón se moviliza de manera similar por la acción de la fosforilasa del almidón. Ambas reacciones liberan glucosa 1- fosfato a partir de los extremos no reductores del polímero de glucosa. La reacción de ruptura está ligeramente desfavorecida en condiciones estándar pero las concentraciones intracelulares relativamente elevadas de fosfato inorgánico hacen que esta reacción opere in vivo casi exclusivamente en la dirección de degradación, en vez de en la dirección de síntesis.
Al igual que la alfa-amilasa, las fosforilasas no son capaces de romper más allá de los puntos de ramificación alfa 1-6, de hecho, la ruptura se detiene a los cuatro residuos de glucosa de un punto de ramificación. El proceso desramificador requiere la acción de una segunda enzima llamada "desramificante" (alfa1-4 glucantransferasa), la cual cataliza dos reacciones. En primer lugar, está la actividad transferasa, en la que la enzima elimina tres de los residuos de glucosa restantes y transfiere este trisacárido intacto al extremo de alguna ramificación externa.
A continuación, el residuo de glucosa que queda unido aún a la cadena por un enlace alfa 1-6 se rompe por la actividad alfa 1-6-glucosidasa, que posee la misma enzima desramificadora. Ello da lugar a una molécula de glucosa libre y una ramificación de tres residuos de glucosa con enlaces alfa 1-4, esta ramificación que ha quedado ahora expuesta puede ser atacada por la fosforilasa. El resultado final de la acción de estas dos enzimas es la degradación completa del glucógeno a glucosa 1- fosfato (el producto final de la glucosa).
La importancia de almacenar energía de los hidratos de carbono en forma de un polímero altamente ramificado puede radicar en la necesidad del animal de generar energía de manera muy rápida, tras los estímulos apropiados. La glucógeno fosforilasa ataca los enlaces exoglucosídicos, es decir, rompe de manera secuencial a partir de los extremos no reductores. Cuantos más extremos de este tipo existen en un polímero con mayor rapidez puede movilizarse.
Para metabolizarse mediante la glucólisis, la glucosa 1- fosfato producida por la acción de la fosforilasa debe convertirse en glucosa 6-fosfato. Esta isomerización la lleva a cabo la fosfoglucomutasa. Desde el punto de vista de su mecanismo, esta reacción es similar a la de la fosfoglicerato mutasa, excepto que en la fosfoglucomutasa hay una fosfoserina en vez de una fosfohistidina en la enzima que reacciona con el sustrato.
La serina que lleva el grupo fosfato es excepcionalmente reactiva, como indica el hecho de que la fosfoglucomutasa, como la quimotripsina y otras proteasas de serina, se inhibe de forma irreversible por el diisopropilfluorofosfato.
Ambos procesos dan lugar a formas fosforiladas de glucosa,que no pueden salir de las células hepáticas. La conversión de glucosa libre se realiza mediante la acción de la glucosa –6-fosfatasa, que hidroliza la glucosa-6.fosfato a glucosa y ortofosfato. Esta enzima está presente también en el riñón y en el intestino. En cambio, el glucógeno muscular se utiliza principalmente como fuente de glucosa-6-fosfato para el catabolismo en las células musculares.
En consecuencia, en el músculo no hay glucosa-6-fosfatasa, como tampoco la hay en el cerebro, que depende casi exclusivamente de la glucosa de la sangre como principal fuente de energía. Ello garantiza que la glucosa-6-fosfato formada a partir del glucógeno no difunda hacia el exterior de estas células, puesto que, como se ha indicado antes, los azúcares fosfato no atraviesan con facilidad las membranas celulares.(7)
1.3 Síntesis del Glucógeno Hepático:

(6) Lynch,M.J., Rápale, S.S., Mellor, L.D., Spare, P.D., Inwood, M.J.H. "Métodos de Laboratorio" 1991. Tercera edición. Editorial: Interamericana, México,D.F. Página 554
La síntesis de glucógeno tiene lugar durante la fase posprandial de absorción, cuando la concentración de glucosa en la vena porta es superior a 150 mg/100ml, y en general cerca de 180mg/100ml durante la absorción activa. Se cree que no hay barrera para la entrada libre de glucosa a los hepatocitos; durante dicha fase de absorción, entran pues a las células del hígado grandes cantidades de glucosa. Este fenómeno inicia la síntesis de la enzima hepática específica de la fosforilación de glucosa llamada glucocinasa. Lo mismo hace la insulina, en tanto que el ayuno o la falta de insulina detienen la síntesis de glucocinasa. Sin embargo, la insulina desencadena un fenómeno de competición por parte de los músculos estriados, pues acelera la entrada de glucosa a las células de éstos, donde interviene en la fabricación de glucógeno. Es probable que, mientras se sintetiza glucocinasa, la hexocinasa inespecífica fosforila la glucosa en el hígado.
Pero el papel en conjunto desempeñado por la hexocinasa es mucho menor que el de la glucocinasa. Ambas enzimas transfieren fosfato del ATP al carbono 6 de la glucosa, pero el producto final (glucosa 6-fosfato) inhibe la hexocinasa. Mientras dura la absorción, la elevada cantidad de glucosa significa un alto nivel de glucocinasa, que forma bastante glucosa 6-fosfato para invertir el equilibrio habitual entre glucosa-1-fosfato y glucosa-6-fosfato, normalmente de 19 a 1, que corresponde a la enzima fosfoglucomutasa. Cuando se ha formado suficiente glucosa 6-fosfato para invertir este ingrediente, se inicia la glucogénesis, apareciendo glucosa 1-fosfato.
Luego la enzima transferasa de uridilo une el trifosfato de uridina a la glucosa-1-fosfato, formando difosfato de uridina y glucosa (UDPG). La unidad glucosilo del UDPG pasa enseguida al glucógeno, al que se une por un enlace alfa-1-4 por acción de la enzima sintetasa de glucógeno (llamada también transglucosilasa UDPG-glucógeno).
En particular, la sintetasa de glucógeno se activa por desfosforilación, mientras que ocurre lo contrario en el caso de la fosforilasa. Tal vez existan relaciones mutuas entre la fosforilación y desfosforilación de estas dos enzimas, de manera que al activarse una, se inactiva otra, y viceversa. La activación de sintetasa de glucógeno aumenta por efecto de la insulina.
La sintetasa de glucógeno fija unidades de glucosilo a las ramas externas del glucógeno, mediante enlaces 1,4 únicamente. Después de añadir de esta manera de 7 a 21 unidades de glucosilo, otra enzima, llamada "de ramificación" (transglucosidasa de amilo 1, 4, 1,6) transfiere algunas de estas unidades y las dispone como ramificaciones, mediante enlaces 1,6 sobre la misma cadena o sobre otra. De esta manera, se obtiene una molécula compacta ramificada; de no ser así, se obtendría una forma alargada, desparramada, parecida a la de un sauce llorón.
Sin embargo, Hers (1964) señala que en el organismo intacto las concentraciones relativas de fosfato inorgánico y de glucosa-1-fosfato de glucosa son tales que la fosforilasa sólo desdobla el glucógeno, y su función de síntesis se traduce mas bien por remodelado de la molécula.
2. Catabolismo del Glucógeno (glucogenólisis):
La glucogenólisis aumenta en el músculo varios cientos de veces inmediatamente después del comienzo de la contracción. Esto comprende la activación rápida de la fosforilasa causada por la activación rápida de la fosforilasa cinasa por el calcio, la misma señal que inicia la contracción. La fosforilasa cinasa muscular tiene cuatro tipos de subunidades: alfa, beta gamma y delta, en una estructura representada como (alfa-beta gamma-delta).
Las subunidades alfa y beta contienen residuos de serina que son fosforilados por la proteincinasa dependiente de AMPc. La subunidad beta fija 4 iones calcio y es idéntica a la proteína fijadora de calcio, calmodulina. La fijación del calcio activa el sitio catalítico de la subunidad gamma en tanto que la molécula permanece en la configuración b desfosforilada. Sin embargo, la forma a fosforilada sólo es activada en forma total en presencia de calcio. En un hecho significativo que la calmodulina sea análoga en estructura a la TpC, la proteína fijadora de calcio en el músculo. Una segunda molécula de calmodulina o de TpC puede interactuar con la fosforilasa cinasa, aumentando la activación. Por lo tanto la activación de la contracción muscular y de la glucogenólisis son realizadas por la misma proteína fijadora de calcio, que asegura su sincronización (9).
Las enzimas que participan en la glucogenólisis son:
2.1 a) Fosforilasa: Es la enzima más importante para el desdoblamiento del glucógeno. Rompe el enlace 1,4 de la unidad de glucosilo del extremo de una rama o cadena de glucógeno, y cataliza simultáneamente la transferencia del glucosilo liberado a un fosfato inorgánico. De esta manera, la fosforilasa puede desdoblar casi la tercera parte de la molécula de glucógeno en glucosa 1-fosfato. Lo que queda de la molécula de glucógeno después de que la fosforilasa ha ejercido su efecto máximo se llama "dextrina límite".
La fosforilasa hepática se activa por transfosforilación (del ATP), debida a la enzima cinasa de desfosfofosforilasa, en presencia de magnesio. Esta activación es acelerada varias veces por el monofosfato cíclico de adenosina (AMP, o fosfato de 3,5-ribosa-adenina cíclica). El AMP se forma a partir del ATP por efecto de la enzima ciclasa de adenilo, que se encuentra en las membranas celulares. El glucágon y la adrenalina triplican la formación de AMP, por lo tanto, activan así la fosforilasa hepática, lo que explica su potente efecto glucogenolítico.
2.2. b) Fosforilasa del Músculo: Esta enzima difiere de la fosforilasa hepática por varias razones, principalmente porque existe en dos formas, las variedades a y b. La fosforilasa a del músculo (P.M. 495 000) es un dímero de b, y contiene cuatro unidades de fosfato de piridoxal por molécula, en tanto que la variedad b sólo contiene dos.
Las dos variedades presentan transformaciones mutuas. En el músculo en reposo predomina ampliamente la fosforilasa b; se activa y convierte en fosforilasa a por efecto de la cinasa de fosforilasa b, activada a su vez por el AMP cíclico. Puesto que la adrenalina (pero no el glucágon) aumenta considerablemente la formación de la AMP cíclico en el músculo, esta hormona aumenta la actividad de las fosforilasas del músculo e hígado, mientras que la acción del glucágon sólo se ejerce sobre el hígado.
En reposo, el AMP cíclico del músculo no basta para activar la fosforilasa, pero el ejercicio muscular y la anaerobiosis probablemente aumentan localmente la concentración del adenilato cíclico.
2.3 Enzima de desramificación (glucosidasa de 1,6 –amilo):
Puesto que la fosforilasa sólo ataca los enlaces 1,4 glucosídicos, deja de actuar cuando llega a un punto de ramificación. Cori y Larner (1951) dedujeron que la fosforólisis de las principales cadenas externas se detiene a varias unidades glucosílicas de distancia de un punto de ramificación; pero en el caso de las ramas laterales, prosigue hasta que sólo queda la unidad de glucosilo fijada por el enlace 1,6. La molécula de glucógeno "deshojada" o podada por la fosforilasa se llama "dextrina límite".
En este punto, la enzima de desramificación ataca el enlace 1,6 en cuestión, liberando unas moléculas de glucosa por cada punto de ramificación, lo que permite que vuelva a actuar la fosforilasa. En teoría cuando menos, estas intervenciones sucesivas de fosforilasa y enzima de desramificación pueden llevar el glucógeno al estado de cadena basal solamente, lo que se podría llamar el tronco; se puede producir así de 92 a 93% de glucosa 1-.fosfato y de 7 a 8% de glucosa libre.
2.4 Glucotranferasa de oligo 1,4 --------1,4:
Walker y Whelan ya sospechaban la presencia de esta enzima como contaminante en los preparados de glucosidasa de 1-6 amilo. En diversos análisis efectuados se obtuvieron resultados que hasta la fecha sugieren que la acción de la fosforilasa sobre las cadenas terminales se detiene a cuatro unidades de glucosilo de distancia de un punto de ramificación. En esta etapa (dextrina límite), la mayor parte de las cadenas externas "desprendidas" de la molécula parecen formadas por cuatro unidades alfa- 1,4-glucosilo, a partir de un punto de ramificación. Se cree que la glucotransferasa de oligo- 1,4 ---------1,4, pasa tres de estas unidades al extremo de otra cadena; por consiguiente, la fosforilasa puede volver a actuar sobre la cadena, ya más larga, y la enzima de desramificación puede atacar el enlace 1,6 en el punto de ramificación.
2.5 Alfa amilasas:
Olivarría y Torres (1962) demostraron que la alfa-amilasa del hígado podía atacar el glucógeno mediante: 1) Producción de oligosacáridos de cadena recta, como maltotriosa y maltotetrosa, a partir de las ramas externas del glucógeno, y 2) Liberación de sacáridos ramificados y de maltosa, desde el interior de la molécula. Existen varias maltasas (glucosidasas alfa 1,4) para transformar estos productos en glucosa. Sin embargo, todavía se ignora la importancia de la vía alfa-amilasa-oligosacárido maltasa en el catabolismo del glucógeno.
2.6 Glucógeno de lisosomas:
Los lisosomas poseen un conjunto de enzimas capaces de hidrolizar prácticamente cualquier componente del citoplasma que contienen entre otras, fosfatasa ácida, RNAasa, DNAasa, catepsina, beta-glucoronidasa, sulfatasa de arilo, beta-N-acetilglucosaminidasa, beta-galactosidasa y alfa-1,4(glucosidasa).
La función de esta última enzima en el metabolismo celular normal no se conoce todavía, pero la falta de maltasa ácida de lisosomas en la enfermedad de Pompe tiene como consecuencia el almacenamiento de glucógeno en acúmulos limitados por membranas (lisosomas). (6)
3. Cascada amplificadora de la degradación del glucógeno, estimulada por adrenalina:
Este proceso se inicia cuando la adrenalina estimula la degradación del glucógeno en el hígado para convertirse a glucosa, originando una serie de reacciones de amplificación (cascada amplificadora), con lo cual se eleva la concentración de glucosa sanguínea. El mecanismo se lleva a cabo como sigue:

(5) Lehninger,Albert L. "Bioquímica. Las Bases Moleculares de la Estructura y Función Celular". 1991. 2da. Edición. Editorial: Ediciones Omega S,A. Barcelona. Página 824
La adrenalina que llega a la superficie de la célula hepática en una concentración de
10-8 a 10-10 M, se une a los centros receptores específicos de la adrenalina de la superficie exterior de la membrana de las células hepáticas. Se cree que esta unión provoca un cambio de conformación local en la membrana que origina la activación de la adenilato-ciclasa localizada sobre la superficie interior de la membrana celular. La forma activa de la adenilato-ciclasa convierte al ATP en AMP cíclico, que puede llegar hasta una concentración cumbre de 10-6 M en el interior celular.
El AMP cíclico así formado se une entonces a la subunidad reguladora de la proteína-quinasa, liberando a su subunidad catalítica en una forma activa. La subunidad cataliza a continuación, la fosforilación de la forma inactiva de la fosforilasa-quinasa a expensas del ATP, para producir la fosforilasa-quinasa activa. Esta enzima, que requiere de calcio para su actividad, cataliza entonces la fosforilación de la fosforilasa b inactiva a expensas del ATP, rindiendo fosforilasa a activa, la cual, a su vez, provoca la degradación catalítica del glucógeno a glucosa-1-fosfato a partir de la cual, se forma glucosa 6-fosfato y después, la glucosa libre de la sangre. Cada una de las etapas de esta cascada es catalítica, y su conjunto acaba en una gran amplificación de la señal que ingresa, esto es, en la unión a la superficie del hepatocito de un número relativamente pequeño de moléculas de adrenalina. A pesar de que la cascada consta de muchas etapas de unas enzimas actuando sobre otros, puede alcanzar su máxima actividad en cuestión de segundos. La adrenalina no sólo estimula la degradación de glucógeno, sino que también inhibe la síntesis del mismo en el hígado, digiriendo así todos los restos de glucosa disponibles y sus precursores hacia la producción de glucosa libre.
La fijación de la adrenalina a la célula hepática y la subsiguiente formación de AMP cíclico promueve la fosforilación por la proteína-quinasa de la forma activa, o de desfosfo, de la glucógeno-sintasa, transformándola en la forma inactiva fosforilada. Seis restos de serina de la molécula de glucógeno-sintasa se fosforilan a expensas del ATP. Así, la inhibición de la glucógeno-sintasa se verifica por una cadena de fenómenos "disparados" o desencadenados por el mismo estímulo que provoca la aceleración de la degradación de glucógeno a glucosa sanguínea. Mientras se secreta adrenalina a la sangre por parte de la médula suprarrenal, el sistema hepático de la adenilato-ciclasa permanece activado, manteniendo el AMP cíclico a gran concentración. Sin embargo, en cuanto la secreción de adrenalina, ésta de detiene la adrenalina unida a la membrana de la célula se desliga. Entonces ya no se forma AMP cíclico, y el restante es destruido por la fosfodiesterasa. Inmediatamente las subunidades de la proteína-quinasa se reasocian formando un complejo que no tiene actividad catalítica. La forma fosforilada de la fosforilasa-quinasa experimenta desfosforilación, lo mismo que la propia fosforilasa a, por la acción de la fosforilasa-fosfatasa. De esta suerte el sistema glucogenolítico es devuelto a su estado normal de reposo; simultáneamente, la glucógeno-sintasa es reactivada por desfosforilación. Aparte de su actividad en el hígado, la adrenalina provoca la degradación de glucógeno en el músculo esquelético, con formación de lactato, gracias a la estimulación de la glucógeno-fosforilasa vía AMP cíclico.
La adrenalina también estimula una lipasa de las células adiposas que degrada escindiendo los triglicéridos, liberando ácidos grasos ligados a la seroalbúmina. Este efecto se verifica por estimulación de la adenilato-ciclasa y por formación de la proteína-quinasa activa, la cual, fosforila al parecer a un precursor inactiva de la lipasa activa de los triglicéridos. Por otra parte, los característicos efectos de la adrenalina sobre el ritmo y el rendimiento cardiacos son mediados por receptores catecolamínicos específicos, así como por la formación de AMP cíclico. Sin embargo, el glucógeno del corazón no se convierte en glucosa sanguínea, sino lactato. El músculo cardiaco y el esquelético carecen de glucosa-6-fosfatasa.
4. Funciones de las reservas de glucógeno en el músculo y el hígado:
El glucógeno constituye la principal fuente de energía para la contracción del músculo esquelético. Dado que el hígado obtiene la mayor parte de su energía metabólica de la oxidación de los ácidos grasos, el glucógeno hepático tiene una función muy distinta, como fuente de glucosa sanguínea que se transporta a otros tejidos para su catabolismo.
El hígado actúa fundamentalmente como un "glucostato", sensor de las concentraciones de glucosa sanguínea que ajusta en función de ello la síntesis y degradación de glucógeno; gran parte de esta regulación comporta el control de la glucógeno sintasa y la fosforilasa. Para cumplir esta función, el hígado contiene unas reservas de glucógeno relativamente elevadas, desde un 2 a un 8% del peso del órgano. En el hígado, la velocidad máxima de síntesis y degradación de glucógeno son aproximadamente iguales, mientras que en el músculo la velocidad máxima de glucogenólisis supera a la síntesis de glucógeno en unas 300 veces. Aunque la enzimología de la síntesis y degradación del glucógeno es semejante en el hígado y el músculo, el control endocrino del hígado es bastante diferente. Las enzimas difieren también estructuralmente. (7)
5. Fisiopatologías del metabolismo del glucógeno:
Las enfermedades relacionadas con el metabolismo del glucógeno se denominan glucogenosis. Se han descrito deficiencias congénitas de la mayoría de las enzimas o transportadores del metabolismo de glucógeno. En el caso de la fosforilasa, se han descrito deficiencias que afectan a la enzima hepática o muscular ya que se trata de proteínas diferentes. Además, cuando son varios los polipéptidos que forman una enzima , cada subunidad puede estar afectada específicamente. Ello explica la existencia de formas ligadas al cromosoma X y una forma autosómica de deficiencia de fosforilasa b quinasa. En 1954, Cori inició la numeración de estas enfermedades, de las que actualmente se conocen hasta la IX. La multiplicidad de defectos que se han descrito, demuestra que el glucógeno no es esencial para la vida.
Por otra parte, está claro que las enfermedades debidas a alteraciones enzimáticas diferentes pueden cursar con manifestaciones clínicas similares. Los órganos más afectados por las alteraciones de las enzimas y el metabolismo del glucógeno son el hígado y el músculo esquelético, dado que es en ellos donde se almacena en una mayor proporción. Si la deficiencia afecta a una enzima hepática (glucogenosis tipos I, III, VI Y VIII) la sintomatología está relacionada con la aparición de hipoglucemia, debido a la incapacidad del hígado para liberar glucosa a partir de glucógeno. Al mismo tiempo aparece hepatomegalia y los hepatocitos adquieren una apariencia arbórea. También es característico que los pacientes no respondan a la administración de glucagón, como hormona hiperglucemiante. Los signos y síntomas clínicos aparecen normalmente en estos pacientes entre el mes y el año de vida y los más característicos son: hipoglucemia, hepatomegalia, cara de muñeca, acidosis láctica, hipertrigliceridemia, hipercolesterolemia, pruebas hepáticas anormales y osteoporosis. Cuando la deficiencia enzimática afecta al músculo (tipos V Y VII), los síntomas clínicos están relacionados con la incapacidad de suministrar combustible metabólico rápido para la contracción muscular. Los síntomas son débiles y sólo son aparentes en el joven al realizar ejercicios violentos. En otras formas de glucogenosis (tipos II Y IV), los problemas están relacionados con el acúmulo de glucógeno en un compartimiento subcelular anormal (tipo II) o con una estructura anormal (tipo IV). (4,6,7,8,10)
5.1 Glucogenosis tipo I: Deficiencia de glucosa 6-fosfatasa (Enfermedad de von Gierke; glucogenosis hepatorrenal):
Se da el nombre del investigador mencionado a esta variedad, aunque no es seguro que el caso estudiado por Von Gierke (1929) haya correspondido a este tipo. Los primeros en demostrar una deficiencia de glucosa6-fosfatasa en estos enfermos fueron Cori y Cori en 1952. Todavía no se conoce la frecuencia exacta pero quizá el tipo I represente casi 25% de todos los casos de glucogenosis. La deficiencia se hereda con carácter autosómico recesivo simple; o sea, los padres de los enfermos son heterocigotos y pueden resultar afectados otros hermanos. (3,4,6)
5.1.1 Manifestaciones clínicas:
Suele existir una gran hepatomegalia, especialmente en los niños, a veces desde el nacimiento. En general, también hay hipertrofia de los riñones, aunque como regla, queda enmascarada por hepatomegalia. Pueden presentarse signos de hipoglucemia grave en las primeras horas o días de la vida, o en época más tardía; a veces no ocurren nunca. Otra manifestación inicial relativamente frecuente es una grave acidosis en las primeras horas o días. Al parecer, algunas familias que presentan este trastorno tienden a manifestar síntomas de hipoglucemia y otras no. Pero en general, los niños y lactantes con glucogenosis de tipo I muestran una notable "resistencia" o "falta de sensibilidad" a la hipoglucemia., incluso se llegó a decir que "se acostumbraban a cifras bajas de glucosa en sangre". Quizá la explicación de esta observación sea que los cerebros de estos enfermos utilizan algún substrato distinto de la glucosa. El crecimiento del pelo es lento, pero se conservan las proporciones entre las distintas partes del cuerpo y el niño va alcanzando con cierto retraso etapas normales del desarrrollo. El trastorno del crecimiento se puede deber a la utilización de ácidos aminados para la formación de glucosa, pues la hipoglucemia constituye un estímulo casi constante para la gluconeogénesis. La hipoglucemia explica la frecuencia de sudoración excesiva (hipersuprarrenalismo), y puede dar lugar a un hipercorticosuprarrenalismo ligero. Es probable que explique además la tendencia habitual a la obesidad, más notable en mejillas "cara de muñeca", mamas, nalgas y superficies posteriores de brazos y muslos. Los xantomas papulares amarillos anaranjados que pueden aparecer sobre los miembros son manifestaciones secundarias de hiperlipemia. Se ha querido explicar por trombopatía (Hers, 1964) una tendencia relativamente frecuente a formación de hematomas, epistaxis, etc. No es raro un cierto grado de anemia, que no se ha explicado satisfactoriamente; también pueden presentarse bruscas crisis de acidosis graves y a veces mortales. (3,4,6,8,10)
5.1.2 Datos de laboratorio:
Después de un periodo de ayuno (de cuatro a seis horas), estos pacientes suelen mostrar hipoglucemia pronunciada, con cifras sanguíneas de glucosa verdadera entre 50 y 0 mg/100ml. Sintetizan y desdoblan normalmente el glucógeno, pero al no poder producir glucosa libre a partir de glucosa-6-fosfato, 93% de su glucógeno hepático resulta inaprovechable para el mantenimiento de la glucemia. Por la misma razón, la respuesta al glucagon (0.02 a 0.1mg/kg de peso corporal) por vía intravenosa o intramuscular, y a la adrenalina (30 microgramos ó 0.03ml de una dilución al 1 por 1000 por kg) por vía subcutánea, falta por completo, o es menor que la normal (aumento de 50% de la glucemia en ayunas en un plazo de 20 a 30 minutos). Sin embargo, como la enzima de desramificación puede liberar de 7 a 8% del glucógeno almacenado como glucosa libre, los pacientes con enfermedad de Von Gierke que se alimentan bien y no llegan a sufrir periodos de ayuno pueden presentar una respuesta ligera o moderada en los estudios con glucagon o adrenalina. Ambas pruebas producen un aumento considerable de lactato en sangre, pues la glucosa 6-fosfato debido a la glucogenólisis pasa a la vía glucolítica. Esta producción de lactato puede desencadenar la acidosis, descendiendo el pH plasmático y el contenido de bióxido de carbono hasta el punto de que se deba administrar bicarbonato de sodio (2meq/kg). También aumenta el piruvato sanguíneo, pero en menor proporción. Es frecuente la hiperuricemia, y se ha visto que podía producir en estos enfermos un cuadro de gota, atribuible a la menor depuración renal de ácido úrico a consecuencia de la cifra elevada de lactato en sangre. La ingestión frecuente de alimentos tiende a mantener dentro de los límites normales los niveles sanguíneos de lactato, piruvato y ácido úrico, pues en estas condiciones la enzima de desramificación permite conservar cifras de glucemia casi normales. Es habitual la hiperlipidemia. Los lípidos totales suelen encontrarse entre 0.8 y 2g/100ml, y pueden ser mucho más altos. Se elevan por los ácidos grasos libres, triglicéridos y colesterol del plasma.La aparición brusca de acidosis peligrosa en estos pacientes, se debe al aumento de la glucogenolisis, con utilización en el ciclo de Embden-Meyerhoff de la glucosa 6-fosfato resultante, que tiende a ocurrir en caso de inanición. Puede haber acetonemia y acetonuria en estas crisis acidóticas, o no.
Al realizar un hemograma se encuentran las plaquetas aumentadas.(2,6).
5.1.3 Pruebas especiales:
Todas las observaciones antes mencionadas, son inespecíficas. Por ejemplo, la falta de respuesta normal al glucágon o la adrenalina podría deberse al agotamiento del glucógeno hepático, o a una deficiencia de fosforilasa o de enzimas de desramificación.
- Prueba de tolerancia a la galactosa. La galactosa se transforma rápidamente en glucosa 1-fosfato; éste, en un sujeto en ayunas, da lugar a glucosa sanguínea libre, en presencia de una fosfoglucomutasa y una glucosa- 6- (fosfatasa) normales. Se realiza la prueba después de cuatro a seis horas de ayuno, por inyección intravenosa de 1.0 g de galactosa por kg. De peso, bajo la forma de solución al 25%, en dos o tres minutos. Se toman muestras de sangre antes de la inyección y a los 10, 20, 30, 40, 50 y 60 minutos de ésta. En los sujetos normales, la glucosa en sangre empieza a aumentar a los 10 minutos de la inyección, y casi no cambia el lactato sanguíneo. En la deficiencia de glucosa-6-fosfatasa, la glucosa sanguínea no aumenta después de la inyección de galactosa, pero el lactato en sangre se eleva mucho en 20 a 30 minutos.
- Prueba de la fructosa de Hers y Malbrain: Se basa en el mismo principio que la prueba de Schwartz. Sin embargo, en teoría, la prueba de la fructosa debería constituir un mejor índice, pues este se transforma en glucosa 6-fosfato, sin que intervenga la fosfoglucomutasa. Pero en la práctica no se conocen casos relacionados con esta particularidad, pues una deficiencia de fosfoglucomutasa, que desempeña, en la síntesis y el desdoblamiento de glucógeno, funciones de "placa giratoria", con toda probabilidad resultaría incompatible con la vida. La técnica de la prueba de la fructosa es igual que la mencionada para la galactosa, pero la dosis es de 0.5 g de fructosa por Kg de peso corporal. Sin embargo, la prueba de la fructosa podría entrañar un mayor peligro de acidosis, tal vez por la utilización brusca de una gran cantidad de glucosa 6- fosfato por la vía de la glucólisis. (1,2,4,6).
En la enfermedad de von Gierke , puede observarse una respuesta hiperglucémica exagerada, con normalización tardía, en las pruebas de tolerancia a la glucosa. Muchas veces resulta bajo el fosfato inorgánico del suero, lo que junto, con la acidosis, podría explicar en parte la observación frecuente de una leve falta de osificación del esqueleto. Una gran infiltración de glucógeno en las células epiteliales del túbulo renal (síndrome de tipo Arman-Ebstein) podría producir cierto grado de aminoaciduria inespecífica. (6).
5.1.5 Diagnóstico específico:
Sólo se puede establecer por estudios enzimáticos sobre cualquiera de los tres tejidos en los cuales existe normalmente glucosa-6-fosfatasa: hígado, riñón y mucosa del intestino delgado. Con mucho, resulta preferible el hígado. Basta una punción biopsia bien hecha en la medición de la glucosa-6-fosfatasa, pero para un análisis completo se requiere casi 1.0g de tejido. En condiciones ideales, debería estudiarse también una biopsia de músculo en todas las glucogenosis. Se quita el exceso de sangre de las muestras tocándolas con un disco de papel filtro. Nunca debe ponerse en solución alguna una muestra de tejido destinada al estudio de glucógeno o enzimas relacionadas; tampoco se debe lavar, ni siquiera en solución salina.Las muestras de biopsia se colocan sin tardanza en un recipiente de vidrio seco, limpio, hermético, y se congelan de inmediato con hielo seco o nitrógeno líquido. En las muestras congeladas, es posible estudiar las enzimas varias semanas o meses más tarde si existe el necesario, se pueden fijar las muestras para estudios de histoquímica y de microscopía electrónica; además, se pueden realizar cortes sobre tejido fresco, para el estudio de la glucosa. El hígado contiene de 5 a 10% de su peso húmedo de glucógeno, y esta sustancia no alcanza niveles tan altos como en la dextrinosis límite. El constante estímulo para la gluconeogénesis, incluyendo el desdoblamiento del glucógeno a lactato, quizá explique la composición de glucógeno en hígado, a pesar de la hipoglucemia y del mayor estímulo para la glucogenolisis en estas condiciones. Además la glucosa-6- fosfato estimula la sintetasa de glucógeno, aunque no se sabe con exactitud si existe un exceso de glucosa- 6- fosfato en los hígados de los pacientes con enfermedad de von Gierke. En general, aumenta el contenido de grasa del hígado, y puede alcanzar hasta 8% del peso húmedo: esto se atribuye a mayor lipogénesis. Las biopsias muestras a veces células llenas de grasas, pero la mayor parte de los hepatocitos presentan una vacuolación "fenestrada" de glucógeno. La composición y estructura del glucógeno existente son normales. (1,6).
5.2 Glucogenosis de tipo II: Falta de maltasa ácida (Deficiencia de alfa-1,4-glucosidasa ácida; enfermedad de Pompe; enfermedad de almacenamiento de glucógeno lisosómico; glucogenosis generalizada):
La glucogenosis tipo II se debe a la deficiencia de la alfa-glucosidasa ácida, lo que origina el acúmulo de glucógeno en vacuolas derivadas de los lisosomas en casi todos los tejidos. Este fue el primero de los errores metabólicos congénitos lisosómicos descritos. La existencia de cardiomegalia, que se ha considerado como una de sus características, no está presente siempre. Aunque prácticamente todas las células se afectan, las alteraciones funcionales son más patentes en el corazón, el músculo esquelético y el sistema nervioso. Aparecen varias formas: infantil, juvenil y adulta. (3,4,6)
5.2.1 Manifestaciones clínicas:
Los niños que sufren enfermedad de Pompe suelen ser normales al nacer; pero casi siempre muestran signos patológicos entre el primero y el quinto mes de la vida, y con pocas excepciones, el desenlace fatal sobreviene en el primer año. La acumulación progresiva de glucógeno en los músculos produce debilidad cada vez mayor, hasta sospechar amiotonía congénita. Por las mismas razones puede presentarse macroglosia, que junto con las alteraciones del sistema nerviosos central, contribuye a dar la impresión de deficiencia mental. El llenado físico por glucógeno de las células musculares explica la debilidad, que puede aumentar a consecuencia de anomalías neuronales. La debilidad de los músculos respiratorios explican la diseña y la cianosis, y finalmente la bronconeumonía, que con frecuencia es causa de muerte. Aunque suele observarse cierto grado de cardiomegalia (la víscera puede tener un tamaño de dos a cinco veces superior al normal) se conocen casos en los cuales el corazón no había crecido. Cuando existe cardiomegalia, no se acompaña de soplos ni otros signos, aunque conduce generalmente a una muerte temprana por una combinación de bronconeumonía e insuficiencia cardiorrespiratoria. El hígado crece ligera o moderadamente, y a veces escapa al examen clínico. (4,6).
5.2.2 Características bioquímicas:
Las mediciones químicas en sangre, como glucosa, tolerancia a la glucosa, lactato, piruvato, lípidos, pH, acetona, respuesta al glucágon y adrenalina, y pruebas con galactosa y fructosa intravenosas, son normales. La tinción con ácido peryódico-Schiff
de frotis de sangre periférica fijados con alcohol permite observar grandes cantidades de glucógeno en los leucocitos. En un caso se demostró que no existía alfa-glucosidasa en los leucocitos.
Entre otros análisis encontramos:
- Hemograma.- Cifras normales, pero leucocitos cargados de glucógeno.
- Química hemática.- No hay alteración de glucemia basal ni tras sobrecarga. CPK disminuida.
- Orina.- Cetonuria negativa.
- Biopsia muscular.- Hallazgo típico, especialmente en la histoquímica, déficit de maltasa ácida. (1,4,6).
En 1963, estudiando tejidos que procedían de cinco pacientes, Hers demostró que faltaba la alfa-glucosidasa ácida en hígado, corazón y músculo estriado, en ese mismo año, Lejeune y colaboradores demostraron que esta enzima, que hidroliza la maltosa, los oligosacáridos de cadena recta y las cadenas externas del glucógeno hasta producir glucosa, y cuyo pH óptimo es de 4, se encontraba en los lisosomas. Luego, el grupo de Louvain demostró que en la enfermedad de Pompe, el almacenamiento excesivo de glucógeno corresponde, en todas las células, a vesículas citoplásmicas limitadas por membranas únicas, de forma irregular y tamaño variable (hasta 8 micras), que interpretaron como lisosomas deformados. Este interesantísimo descubrimiento no explica por completo la naturaleza de la enfermedad de Pompe. Quizá los lisosomas "engullen" normalmente glucógeno, junto con organelos citoplásmicos gastados o sobrantes, y en la enfermedad de Pompe se acumule gradualmente glucógeno en ellos. Sin embargo, esto requiere además que los lisosomas normales contengan alfa-1,6-.glucosidasa, pues la maltasa ácida por sí sola no bastaría para desdoblar completamente el glucógeno. Además, no se explica tampoco el aumento de 500% de la actividad de fosfatasa ácida en el hígado y el músculo de estos enfermos. El contenido tisular de glucógeno suele ser mayor en la enfermedad de Pompe que en cualquiera otra glucogenosis. Hers (1955) encontró valores medios (respecto a peso húmedo) de contenido de glucógeno en estos enfermos de músculo estriado 11%, músculo cardiaco 6.5%, hígado 9%. En el hígado, resultan afectados tanto los hepatocitos como las células de Kupffer, y en las preparaciones habituales las vacuolas de glucógeno tienen un aspecto más neto, regular, redondo, de tipo gota de grasa, que en cualquiera otra variedad de enfermedad por almacenamiento de glucógeno. Los cortes de músculo casi no permiten identificar el tejido, y sólo muestran miofibrillas muy separadas, en una red longitudinal de vacuolas. Con cierta frecuencia existe también una substancia basófila, que parece ser un mucopolisacárido ácido. Las neuronas pueden encontrarse muy hinchadas, y en las preparaciones habituales no es posible distinguirlas de las que se observan en el caso de enfermedades de Niemann-Pick o Tay-Sachs. En la enfermedad de Pompe, no hay acumulación de glucógeno en los núcleos. (2,6).
5.3 Glucogenosis tipo III: Deficiencia de amilo 1,6-glucosidasa (Enfermedad de Forbes; deficiencia de la desramificación : dextrinosis límite):
Este tipo de glucogenosis aparece como consecuencia de la deficiencia de la enzima desramificante y se le conoce también como dextrinosis límite o enfermedad de Forbes. Se caracteriza por la existencia de un glucógeno de estructura anormal que se acumula en exceso en el hígado y en los músculos. Durante los primeros años es difícil distinguirla de una glucogenosis tipo I pero con la edad la hepatomegalia llega incluso a desaparecer. Los síntomas musculares aparecen en la edad adulta y no en todos los pacientes. En 1953 Forbes estudió una niña de 12 años y medio con una enfermedad de almacenamiento de glucógeno en la cual el glucógeno era anormal, en el sentido de mostrar ramas externas muy cortas. Pensó en una deficiencia de enzima de desramificación. (3,4,6).
5.3.1 Manifestaciones clínicas:
Clínicamente, esta enfermedad recuerda los casos leves de deficiencia de glucosa-6-fosfatasa (tipo I). Se observan hipoglucemia, acidosis, hiperlipemia y retraso del crecimiento, pero ligeros. En los pacientes de tipo Forbes, la glucemia en ayunas se encuentra entre 34 y 56mg/100ml, y el colesterol sérico es vecino de 280 mg/100ml. Las biopsias de hígado efectuadas cuando la enferma tenía 12 años de edad, mostraron aumento de grasas y cierta proliferación del tejido fibrorreticular. La hepatomegalia es generalmente muy notable, pero tiende a disminuir en la adolescencia. Al pasar el tiempo, también, la dextrinosis límite parece hacerse menos grave, y probablemente es compatible con una vida de duración normal. (4,6,8,10).
5.3.2 Datos de laboratorio:
Se observa una leve hipoglucemia en ayunas, y el lactato sanguíneo no es tan alto como en los pacientes de tipo I. La respuesta al glucágon o la adrenalina después de un ayuno breve (de cuatro a seis horas) puede ser un poco menor que la normal solamente, pero después de un ayuno de toda la noche (de 12 a 14 horas), las respuesta es muy pequeña o casi nula. En las pruebas con galactosa o fructosa intravenosa, la respuesta es normal.
(1,2,4,6).
5.3.3 Anatomía patológica:
Se depositan grandes cantidades de glucógeno en el hígado (a veces más de 10% del peso húmedo del órgano) y un poco menos en músculos (generalmente entre 3 y 4%, y nunca más de 8%). Si se recoge glucógeno y se analiza, inmediatamente después de la fase de absorción, los resultados son normales; pero el glucógeno recogido después de 12 horas de ayuno muestra cadenas externas muy cortas, con un aumento de 40 a 50% del número de puntos de ramificación en relación con la estructura normal ( de 7 a 8%). O sea, en estado de ayuno, estos pacientes muestran reducción del glucógeno al estado de dextrina límite, después de lo cual no se puede desdoblar ya, por falta de enzimas de desramificación. El diagnóstico definitivo requiere estudios enzimáticos de hígado y músculo. William y col. (1963) han descrito una deficiencia de enzima de desramificación en los leucocitos de estos enfermos. (8,6).
5.4 Glucogenosis tipo IV: Deficiencia de glucosidasa de amilo-1,4-1,6 (Enfermedad de Andersen, amilopectinosis, enfermedad por falta de ramificación):
Esta variedad es muy rara, y sólo se han descrito dos casos hasta la fecha. El enfermo de Andersen (1956) era un niño de sexo masculino de 11 meses, que mostraba hepatomegalia y ascitis, y murió a los 17 meses. En las células del hígado y del sistema reticuloendotelial, se encontraba un glucógeno anormal, muy poco soluble en agua, que daba reacciones de tipo amilopectina con el yodo. Analizando este glucógeno, G. T.Cori encontró para las cadenas internas y externas una longitud media de 21 unidades de glucosilo (longitud normal de 11 a 13). A partir de esta observación, Cori pensó en una deficiencia de enzima de ramificación (no disponía de tejido para estudios enzimáticos). En forma sorprendente, el hígado sólo contenía 2.8% de glucógeno, y no había exceso de esta sustancia en el músculo. No se sabe por qué una cantidad relativamente tan pequeña de glucógeno anormal puede producir cirrosis hepática, pero se piensa en una acción irritante debida a la falta de solubilidad. La presencia de glucógeno anormal en células reticuloendoteliales sugiere fagocitosis a partir del plasma, pero se requeriría además deficiencia de alfa-amilasa del plasma. Aunque se desconoce la base genética de esta enfermedad, un hermano del enfermo estudiado por Andersen había muerto antes con un cuadro semejante. (3,4,6).
5.4.1 Datos de laboratorio:
*Hemograma.- Leucocitosis ocasional. Carencia de amilo-1,4-1,6-transglucosidasa en los leucocitos. Anemia discreta.
*Química hemática.-Curva de glucemia plana tras adrenalina o glucagón.
*Pruebas funcionales hepáticas.- A menudo alteradas
*Biopsia hepática.- Típica, glucógeno anormal amilopectoideo. Déficit de la enzima circulante. (1,2)
5.5 Glucogenosis tipo V: Deficiencia de miofosforilasa (Enfermedad de McArdle):
La primera observación sobre esta enfermedad fue realizada por McArdle en un joven que tras realizar ejercicio suave mostraba fuertes dolores, debilidad y rigidez muscular. Por el contrario, el lactado sanguíneo en lugar de elevarse, como es lo normal, descendía y su elevación en respuesta a la adrenalina era inferior a la normal. Estos hechos llevaron a sospechar que el paciente no podía convertir el glucógeno en lactato, demostrándose posteriormente que existía una deficiencia de fosforilasa muscular. Por el contrario, el hígado es normal y no aparece hipoglicemia. Las características clínicas principales de la glucogenosis de tipo V son intolerancia al ejercicio, mioglobinuria, fallo renal, debilidad muscular, elevación de la creatina quinasa y electromiograma anormal en reposo. Es frecuente la aparición de calambres y contracturas en la segunda o tercera década de la vida. (3,4)
5.5.1 Datos de laboratorio:
Química hemática.- No aumenta la lactocidemia, ni la piruvicemia por esfuerzo, creatinfosfoquinasa y aldolasa elevadas.
Orina.- Mioglobinuria de esfuerzo.
Biopsia muscular.- Deficiencia de fosforilasa a y b, total o parcial. (1,2,6)
5.6 Glucogenosis tipo VI: Deficiencia de fosforilasa del hígado (Enfermedad de Hers):
Esta enfermedad fue descrita en 1959 por Hers, que había estudiado un año antes tres pacientes con enfermedad hepatomegálica de almacenamiento de glucógeno, viendo que en estos hígados sólo existía 25% de la actividad normal de fosforilasa. Desde entonces, Hers ha estudiado otros muchos casos similares, y concluye que se trata probablemente de enfermedades de almacenamiento de glucógeno; representaría hasta 30 a 35% de todos los casos. Es importante señalar que en ninguno de los pacientes estudiados faltaba por completo la fosforilasa hepática; el nivel más bajo era del orden de 10 a 15% de la cifra normal (3,4)
5.6.1 Fosforilasa de leucocitos:
Se ha encontrado que en los pacientes de tipo VI, es muy baja la actividad de fosforilasa en leucocitos de sangre periférica. También se ha demostrado que las madres de los niños enfermos presentan también una menor actividad de fosforilasa de leucocitos, pero sin manifestaciones patológicas. (6).
5.6.2 Manifestaciones clínicas:
Clínicamente, la glucogenosis de tipo VI es una enfermedad leve, con pronóstico excelente. La hipoglucemia en ayunas generalmente no pasa de 50 mg/100ml. Asimismo, no son muy pronunciadas la cetoacidosis, lacticacidemia, hiperlipemia y efectos sobre el crecimiento. La hepatomegalia es algo más notable, cuando menos en los primeros años. La respuesta al glucágon o la adrenalina es variable (desde una importante deficiencia hasta cifras casi normales). La administración por vía intravenosa de galactosa o fructosa va seguida de una respuesta hiperglucémica normal. La estructura del glucógeno que se almacena es normal también. El diagnóstico definitivo sólo se puede establecer por estudios directos de hígado y músculo. (4)
5.6.3 Datos de laboratorio:
Sangre.- Hiperlipemia con hipercolesterolemia. Cetosis.
Biopsia hepática.- Sobrecarga de glucógeno. Fosforilasa disminuida casi completamente. (1,2).
5.7 Glucogenosis tipo VII (Enfermedad de Tauri):
Esta glucogenosis aparece como consecuencia de la deficiencia de fosfofructoquinasa-1 y es la más rara de todas. La enzima del hígado es normal pero la de los eritrocitos muestra un 50% de la actividad normal. La sintomatología es similar aunque más grave que la de la enfermedad de McArdle, apareciendo también anemia hemolítica. (3,4)
5.8 Glucogenosis tipo IX:
Esta es la única enfermedad en la que existe deficiencia, en lugar de incremento de glucógeno. A pesar de que la actividad sintasa es muy baja en estos individuos, la deficiencia podría no residir en la propia sintasa. (3,4).
5.8.1 Datos de laboratorio:
Bioquímica hemática.- Elevación de la glucemia por glucagon
Biopsia de hígado.- Deficiencia de fosforilasa-quinasa. (1)
RESUMEN DE GLUCOGENOSIS
| TIPO | ENZIMA DEFECTUOSA | ÓRGANO AFECTADO | GLUCÓGENO |
| I | Glucosa-6-fosfatasa | Hígado y Riñón | *Cantidad elevada *Estructura normal |
| II | Alfa-glucosidasa lisosómica | Todos los órganos | *Muy incrementado *Estructura normal |
| III | Enzima desramificante | Músculo e Hígado | *Cantidad elevada *Ramificaciones cortas |
| IV | Enzima ramificante | Hígado y Bazo | *Cantidad normal *Pocas ramificaciones |
| V | Fosforilasa | Músculo | *Cantidad incrementada *Estructura normal |
| VI | Fosforilasa | Hígado | *Cantidad incrementada |
| VII | Fosfofructoquinasa | Músculo | *Cantidad incrementada *Estructura normal |
| VIII | Fosforilasa b quinasa | Hígado | *Cantidad incrementada *Estructura normal |
| IX | Glucógeno Sintasa | Hígado | *Cantidad disminuida |
- Balcells, alfonso. "La clínica y el laboratorio". 1998. 16ava. Edición. Editorial: Masson-Salvat Medicina. Páginas: 517,518.
- Bernard, Henry john. " Diagnóstico y tratamiento clínicos por el laboratorio". 1998. Editorial: Masso-Salvat Medicina. Páginas: 168,290.
- Devlin, Thomas M. "Textbook of Biochemistry with clinical correlations". 1993. Editorial: Willey-Liss. New York 333,334,335,336.
- González de Buitrago, J.M., Arilla Ferreiro, E, Rodríguez-Segade, M. Sánchez Pozo, a. " Bioquímica Clínica". 2000. 1ª. Edición. Editorial: Mc Graw Hill- Interamericana. Páginas: 154,155,156,157,158,159.
- Lehninger,Albert L. "Bioquímica. Las Bases Moleculares de la Estructura y Función Celular". 1991. 2da. Edición. Editorial: Ediciones Omega S,A. Barcelona. Páginas: 272,443,444,445,659,660,661,823,824,825,826.
- Lynch,M.J., Rápale, S.S., Mellor, L.D., Spare, P.D., Inwood, M.J.H. "Métodos de Laboratorio" 1991. Tercera edición. Editorial: Interamericana, México,D.F. Páginas: 553,554,555,556,557,558,559,560,561,562,563.
- Mathews, Christopher K., Van Holde, K.E. "Bioquímica". 1998. 1a. edición. Editorial: Mc Graw Hill –Interamericana. Madrid, España. Páginas: 521,522,523,626,627,628,629,630,631,632,633.
- McPhee, Stephen J., Lingappa, Vishwanath R. Ganong, William, F. , Lange, Jack. D. "Fisiopatología Médica". 1998. 1a. edición. Editorial: El Manual Moderno. Páginas: 155,178 ,214.
- Murray, Robert K., Granner, Daryl K., Mayes, Peter, A. ,Rodwell, Víctor W.F. "Bioquímica de Harper". 1992. 14a. edición. Editorial: El Manual Moderno. Páginas: 132,133,150,164,172,173,174,175,176,177,178,179,647.
- Tórtora, Gerard J. , Anagnostakos, Nicholas, P. "Principios de Anatomía y Fisiología". 1990. 8ª. Edición. Editorial: Harla. Páginas: 281,282 ,283.
- __________________
Recien nacido
Juan es un recien nacido de 6 meses, en esta etapa de la vida existen particularidades biologicas, psicologicas y sociales que van acompañadas de necesidades nutricionales definidas.
Desde el punto de vista psicológico y social, se encuentra en (elegir etapa de la instalacion del psiquismo y describirla brevemente) en esta etapa que se da durante la niñez tienen principal importancia la relacion afectiva entre la marde y el hijo ya que estimula el desarrollo en el niño para establecer las bases de las relaciones objetales que le permiten relacionarse con las cosas. La funcion materna es de mostracion de objetos para estimular al niño a relacionarse con los objetos y representa el factor sociedad. De sostenimiento y de manipulacion porque guia al niño a discriminar entre lo real y lo irreal.
Desde el punto de vista psicológico y social, se encuentra en (elegir etapa de la instalacion del psiquismo y describirla brevemente) en esta etapa que se da durante la niñez tienen principal importancia la relacion afectiva entre la marde y el hijo ya que estimula el desarrollo en el niño para establecer las bases de las relaciones objetales que le permiten relacionarse con las cosas. La funcion materna es de mostracion de objetos para estimular al niño a relacionarse con los objetos y representa el factor sociedad. De sostenimiento y de manipulacion porque guia al niño a discriminar entre lo real y lo irreal.
La comunicación dirigida y transmitida con ayuda de señales y signos ayudara para el desarrollo de la funcion simbolica. Tambien ayudara la resolucion del complejo de edipo correctamente para que la familia no se cierre sobre si misma, dando acceso a la socializacion del niño.
Respecto a lo biologico, se van a producir en el lactante el crecimiento de las estructuras anatómicas en general para su CyD, los reflejos arcaicos constituyen los primeros signos de desarrollo en el niño que luego se iran diferenciando en comportamientos mas específicos.
Para su alimentación tan necesaria deberán estar presentes, el reflejo de succion (…), y el aparato alimentador (…) en correctas condiciones tanto por parte del lactante como de la madre para lograr un acople al pecho adecuado para la lactancia.
En esta etapa el sistema inmunológico del niño se encuentra en desarrollo y puede ser victima de numerosas infecciones por lo que es importante además de una buena alimentación e inmunización que provee la leche materna por sus componentes (IgA y proteínas antibacterianas) y la Ig que atraviesa la barrera placentaria durante el embarazo, llevar un esquema completo de vacunación con el respectivo carnet para controlar las dosis administradas y programar las futuras administraciones. (…)
En cuanto la las necesidades nutricionales, estas guardan relación con los cambios que se generan en su organismo. Ademas de la necesidad de defenderse contra infecciones, se suma la de cubrir los gastos energéticos de su metabolismo basal y actividades diarias, el desarrollo constante del sistema nervioso. Todos cubiertos por la leche materna gracias a su composición en
proteínas (para la formación de tejido y órganos y defensa de MO)
carbohidratos (que proveen la energía necesaria para cubrir los gastos energéticos)
lípidos (escenciales, energéticos y para la maduración del snc)
minerales (Ca y P para el crecimiento y mineralización de huesos y dientes)
y vitaminas (D para la absorción y metabolismo del Ca y Fe)
carbohidratos (que proveen la energía necesaria para cubrir los gastos energéticos)
lípidos (escenciales, energéticos y para la maduración del snc)
minerales (Ca y P para el crecimiento y mineralización de huesos y dientes)
y vitaminas (D para la absorción y metabolismo del Ca y Fe)
Hasta los 6 meses de edad la leche materna cubre las necesidades CyD del lactante, a partir de entonces es necesario introducir una alimentación mixta adecuada para cubrir la demanda nutricional del niño. Ocurren en este período, procesos fisiológicos que brindan aptitud al organismo para recibir estos alimentos. La amilasa pancreática ha madurado para la degradación de hidratos de carbono; la maduración gástrica permite que ya a esta edad no exista reflujo gastroesofágico; existe una madurez intestinal que permite la introducción de nuevos alimentos. La función renal a los 6 meses permite tolerar una mayor carga renal. Comienza a emerger la primera dentadura. En este período el niño ya ha perdido el reflejo de protrusión y comienza con movimientos masticatorios. Puede mantenerse sentado, posición que favorece el inicio de la alimentación semisólida.
En el período comprendido entre el nacimiento y los 2 años, concluye una primera etapa de lactancia materna exclusiva a los 6 meses de edad para iniciar la de la alimentación complementaria hasta los 24 meses, pasando por una etapa de alimentos transicionales hasta la inclusión de los alimentos familiares. Debe llevarse a cabo siempre cumpliendo las 4 leyes para una alimentación saludable y respetando sus sensaciones de hambre y saciedad en un marco de afecto y contención.
Los hábitos alimentarios saludables que se adquieren en la infancia propician buenas condiciones de salud a lo largo de la vida y este es el momento del inicio. La mesa familiar es un momento de comunicación entre quienes la comparten y esto incluye al niño pequeño que se inicia en la alimentación familiar que al sentirse escuchado y comprendido gana seguridad y una buena relación con el acto de comer.
El ritmo de crecimiento en el peso y talla son importantes en la primer infancia 0 a 2a y disminuyen en la sgunda 5 a 10/12ª donde predomina el CyD del sn, social y cognitivo, por tanto disminuye el apetito y puede ser motivo de preocupación por parte de los padres. La masa grasa disminuye de manera gradual.
En este periodo es conveniente darles porciones pequeñas de alimentos varias veces al dia y se debe evitar el consumo de jugos y alimentos azucarados para la protección de las caries dentales y pq reemplazan el consumo de leche y agua. Tambien la alimentación antes de una comida disminuirá el apetito a la hora de la misma.
Debe asegurarse que los padres comprendan cuales son los beneficios de la lactancia y la importancia de una alimentación equilibrada según las 4 leyes formuladas por pedro escudero y las reglas de oro para una alimentación saludable.
PRE ESCOLAR 2-5 a
Juan es un niño de 4 años que se encuentra en el periodo preescolar de la segunda infancia en esta etapa de la vida existen particularidades biologicas, psicologicas y sociales que van acompañadas de necesidades nutricionales definidas.
Desde el punto de vista psicológico y social, el niño se encuentra en la 2da etapa de la conformación de su aparato psíquico, la etapa anal donde su zona erógena y de causa de placer se centran en la retención y expulsión de los excrementos gracias al control que adquieren en esta etapa sobre los esfínteres anal y vesical.
Desde el punto de vista psicológico y social, el niño se encuentra en la 2da etapa de la conformación de su aparato psíquico, la etapa anal donde su zona erógena y de causa de placer se centran en la retención y expulsión de los excrementos gracias al control que adquieren en esta etapa sobre los esfínteres anal y vesical.
Respecto a lo biologico, ocurre una desaceleración del crecimiento corporal, predominando el desarrollo del sn, social y cognitivo. En relación al cese temporal del crecimiento, tb disminuye el apetito.
En cuanto la las necesidades nutricionales, la edad preescolar comprende a los niños de 2 a 5 años. En esta etapa el niño ya está incorporado a la mesa familiar, por lo tanto deberá tener una dieta variada y completa.
En este período existe una desaceleración del crecimiento, por lo tanto el apetito se encuentra disminuido. Puede ser motivo de preocupación por parte de los padres al descubrir que los niños disminuyen el consumo e interés por los alimentos. Para ello es aconsejable darles porciones pequeñas de alimentos varias veces al dia, respetando las sensaciones de hambre y saciedad. Se debe evitar el consumo de jugos y alimentos azucarados para la protección de las caries dentales y pq reemplazan el consumo de leche y agua, a la ves que disminuyen el apetito.
Durante este periodo la masa grasa disminuye gradualmente.
Alrededor de los 3 años, reaparece la neofobia, que coincide con la etapa de rebeldía y del desarrollo del yo. Los reiterados contactos del niño con el alimento, podrán reducirla.
=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/=/
Con respecto a los escolares, que incluye a niñas de 6 a 10 años y a los niños de 6 a 12 años, se lleva a cabo un crecimiento latente, estable y los cambios corporales se producen de manera gradual. Hacia el final de esta etapa comienzan a visualizarse las diferencias por sexo.
Además de ello existe una mayor independencia de su familia producto de los ritmos de la escuela. Pasar más tiempo fuera del hogar conduce a que aumenten sus fuentes de influencia para la selección y hábitos alimentarios.
adolescencia
Juan es un adolescente de 14 años en esta etapa de la vida existen particularidades biologicas, psicologicas y sociales que van acompañadas de necesidades nutricionales definidas.
Desde el punto de vista psicológico y social,
Desde el punto de vista psicológico y social,
Respecto a lo biologico,
En cuanto la las necesidades nutricionales, La adolescencia es el período que se inicia con la aparición de los caracteres sexuales secundarios y concluye con la detención del crecimiento físico; comienza a los 10 años en las mujeres y a los 12 años en el varón, hasta los 18 años aproximadamente en ambos grupos.
Es una etapa en la cual, además de haber importantes cambios físicos, también están presentes los cambios psicosociales, que conllevan a la transición de niño a adulto.
La presión del entorno comienza a imponerse sobre la autoridad de los padres, y eso sumado a los tiempos que corren, conduce a que se vean influenciados sus hábitos y conductas. Puede aparecer la preocupación por la imagen corporal, sobretodo en el sexo femenino, aunque no es un tema ajeno al sexo masculino.
La omisión del desayuno es una conducta reiterada tanto en escolares como en adolescentes; al igual que el sedentarismo y la mala calidad de alimentos que seleccionan.
El plan de alimentación en las etapas que fueron desarrolladas en el capítulo anterior como en la adolescencia debe incluir alimentos de todos los grupos: cereales y legumbres, verduras y frutas, lácteos, carnes y huevo, aceites y grasas, azúcares y dulces. Las proporciones en la dieta diaria se visualizan claramente a continuación:
adulto joven
Juan es un adulto joven de 35 años en esta etapa de la vida existen particularidades biologicas, psicologicas y sociales que van acompañadas de necesidades nutricionales definidas.
Desde el punto de vista psicológico y social,
Desde el punto de vista psicológico y social,
Respecto a lo biologico,
En cuanto la las necesidades nutricionales,
adulto mayor
Juan es un adulto mayor de 70 años en esta etapa de la vida existen particularidades biologicas, psicologicas y sociales que van acompañadas de necesidades nutricionales definidas.
Desde el punto de vista psicológico y social,
Desde el punto de vista psicológico y social,
Respecto a lo biologico,
En cuanto la las necesidades nutricionales,